INTRODUCTION
Adrenocortical carcinoma (ACC) is an extremely rare neoplasm in children and adolescents with an annual incidence of 0.3 cases per million children younger than 15 years old.1) Although the molecular pathogenesis of ACC is poorly understood, it is strongly associated with inactivating mutation of the tumor suppressor gene p53 and alteration of the 11p15 locus leading to insulin-like growth factor 2 overexpression.2) ACC is often associated with the Li-Fraumeni and Beckwith-Wiedemann syndromes.3) Adrenocortical tumors in children and adolescents are frequently hormone-secreting tumors, and virilization alone or in combination with signs of overproduction of adrenocortical hormones is the most common endocrine syndrome.1) Hormonally inactive ACCs, which are common in adults, usually induce abdominal discomfort or back pain caused by the mass effect of the large tumor. Any virilizing symptoms that were resolved after surgical removal of the tumor could theoretically trigger true precocious puberty because of the maturation of the hypothalamic-pituitary-gonadal axis. However, only a few cases have been reported in the world thus far. Therefore, we report a case of virilizing ACC in an 8-year-old boy who subsequently developed central precocious puberty following surgery with reviews of the literature.
CASE REPORT
An 8-year, 2-months-old boy presented to our pediatric clinic with early development of pubic hair and penile growth for the past 3 months. He was born after 40 weeks of gestation through vaginal delivery with a birth weight of 3,600 g. His past medical and surgical histories were unremarkable. A neonatal screening test including 17α-hydroxy progesterone (17-OHP) was negative. None of his family members had endocrine tumors or genetic diseases. On physical examination, his height was 137.6 cm (95th percentile), and his body weight was 40.1 kg (95-97th percentile). His Tanner stage was II. He had no other abnormalities, such as hypertension, cushingoid symptoms, hyperpigmentation, or macroglossia.
The patient was subjected to serum and urinary laboratory tests. Follicle-stimulating hormone (FSH) and luteinizing hormone (LH) levels were below the limit of detection (<0.07 mIU/mL). The levels of serum dehydroepiandrosterone sulfate (DHEAS, 584 µg/dL), 17-OHP (4.05 ng/mL), testosterone (3.82 ng/mL), and urinary 17-ketosteroid (58.8 mg/d) were markedly elevated. The serum morning cortisol level (15.2 µg/dL) was not elevated, and his adrenocorticotropic hormone level was within the normal range (7.89 pg/mL). Meanwhile, his midnight cortisol level (15.0 µg/dL) was not suppressed. In addition, his daily urinary free cortical excretion (106.9 µg/d) was increased. Other laboratory results were within normal limits.
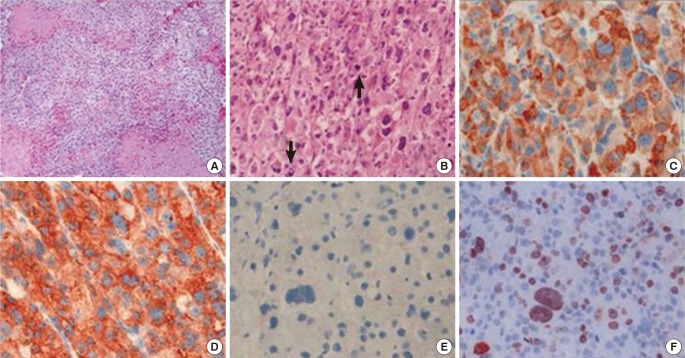
His bone age was 9 to 10 years based on the Greulich-Pyle method. Both abdominal ultrasonography and computed tomography (CT) demonstrated a large right suprarenal heterogeneous mass measuring 7.4×5.7 cm2 with poor enhancement (Figure 1). The lungs, bone, and liver were free from metastases on magnetic resonance imaging or high-resolution CT. A provisional diagnosis of adrenocortical tumor was made, and resection of the right adrenal gland with the tumor mass was performed. Grossly, there was no metastasis to regional lymph nodes or vessels detected during the operation. There was no mutation in the p53 gene. The adrenalectomy specimen was partially encapsulated and soft, measuring 7.8×6.1×4.0 cm3 and weighting 86 g. The cut section was bright brownish with areas of focal hemorrhage. Light microscopic findings of the specimen are shown in Figure 2. The tumor cells were arranged in a nodular and diffuse architecture, and they had high mitotic activity (9/10 high-power fields). Areas of necrosis were present, but capsular or vascular invasion was not observed. The Ki67 labeling index was 30%. According to the criterion of Weiss, a histological diagnosis of ACC was made (Weiss score was 6 of 9). The staging of adrenocortical tumors in this patient (modified from Sandrini et al.) was stage I.2)
After the operation, serum DHEA-S, 17-OHP, and testosterone levels rapidly decreased to the normal levels. However, penis enlargement and the presence of pubic hair persisted at the same level after surgery. Two months after the operation, testicular enlargement and frequent penile erections were noticed by his mother. The volumes of both testes were increased to 5 mL, and his height velocity was increased (10.0 cm per year). The serum DHEA-S level was not elevated, whereas FSH (1.8 mIU/mL), LH (5.9 mIU/mL), and testosterone (3.34 ng/mL) levels were elevated to pubertal levels. The peak values of FSH and LH based on a gonadotropin-releasing hormone (GnRH) stimulation test were at pubertal levels (basal FSH/LH, 2.3/4.4 mIU/mL; peak FSH/LH, 4.5/34.7 mIU/mL), suggesting secondary central precocious puberty. Treatment with a GnRH agonist was initiated, and his pubertal state did not progress further. He remained disease-free approximately 1 year after the operation.
DISCUSSION
ACC is a rare pediatric neoplasm with a poor prognosis. Although an exceptionally high incidence of ACC has been reported for children in southern Brazil and Los Angeles, CA, the worldwide incidence has been estimated at approximately 0.4 cases per million children younger than 4 years of age. ACC has a peak incidence in children younger than 4 years.4) In addition, ACC comprises approximately 0.2% of all childhood neoplasms in the United States according to Surveillance Epidemiology and End Results data from the National Cancer Institute.1,4) A slight female preponderance and a bimodal age distribution have been noted.4) ACC can arise as an either nonfunctional or functional tumor. Non-functional ACC is most common in adults, whereas more than 90% of childhood ACCs are functional.2) Virilization alone or in combination with signs of overproduction is the most common presenting endocrine syndrome in pediatric patients.1) Isolated Cushing's syndrome, the major symptom of adult ACC, is extremely rare, as well as Conn's syndrome.
Our male patient presented with pubic hair, penile growth, and facial acne. His hormonal findings were characterized by the overproduction of androgens in serum and urine analysis. Although his serum DHEA-S levels had remarkably decreased to normal after the operation, the physical signs of precocious puberty were persistent and indicative of progression. In addition, the peaks of FSH and LH in GnRH stimulation testing were at pubertal levels, suggesting secondary central precocious puberty. Several causes of peripheral precocious puberty have been recognized, including adrenal tumors.5) Peripheral precocious puberty may result in the activation of pulsatile GnRH secretion and central precocious puberty. In addition, effective treatment of the primary disease causes the development of secondary central precocious puberty by activating the hypothalamic GnRH pulse generator via a feedback system, as observed in congenital adrenal hyperplasia and familial or sporadic male-limited precocious puberty.6) The period of development for secondary central precocious puberty is various, and other reports illustrated that central precocious puberty may develop as early as 1 month after the initiation of treatment for peripheral precocious puberty.7) GnRH agonists are indicated for the treatment of progressive central precocious puberty, and this therapy was initiated in our male patient to prevent pubertal progressions.
All ACCs are highly malignant tumors, and hence, surgical intervention is the mainstay of treatment for ACC. Because of the friability of these tumors and the potential for tumor spillage, open adrenalectomy is recommended for children with ACC. When metastatic disease cannot be removed or a high risk of recurrence is identified, chemotherapy is often considered. However, the benefits and risks of chemotherapy for ACC in children, including mitotane or other chemotherapeutic agents, have not yet been established.8) Because of the tumor stage in our patient, he was only treated with surgery.
In conclusion, we suggest that in functioning ACCs, clinical follow-up for potential secondary effects of excessive hormone secretion or recurrence after surgical removal is important, and hormonal marker or sexual maturity should be assessed at least every 1 or 2 months after surgery.