INTRODUCTION
Acquired hemophilia is an autoimmune disease characterized by the production of autoantibodies against plasma coagulation factors [1]. It is a rare but potentially life-threatening condition that occurs more commonly in the elderly population [2]. The diagnosis of this condition is under-recognized, and a delay in diagnosis leads to a high mortality rate among these patients. In the elderly group of patients, the unexplained bleeding tendency with isolated prolonged activated partial thromboplastin time (aPTT) should raise high clinical suspicion of acquired hemophilia. Treatment of acquired hemophilia includes acute bleeding control and suppression of the autoimmune process by an immunosuppressive agent [3,4].
CASE REPORT
A 67-year-old man with underlying ischemic heart disease and dyslipidemia presented to the emergency department with spontaneous left leg swelling for a week. There were no complaints of any calf pain or bleeding tendency elsewhere. The patient had no history of preceding trauma or consumption of traditional supplements. Clinical examination revealed a hematoma measuring 10 cmû6 cm in the left calf; however, there was no evidence of hepatosplenomegaly. No other signs of bleeding were noted. The patient did not report of any personal or family history of bleeding disorders. He was compliant with his medications, which included Tab Ramipril (5 mg daily) and Tab Atorvastatin (40 mg, taken at night).
He was initially suspected to have lower left limb deep vein thrombosis; however, the results of D-dimer and ultrasound Doppler tests of the affected limb were found to be negative. The prothrombin ratio and platelet counts were normal but the aPTT was prolonged (55.8 seconds) relative to control (30ã45 seconds). Following his recovery, he was discharged from casualty and referred to primary care for a follow-up examination of the swelling.
During subsequent visits to the clinic, the patient was attended by different doctors; however, his condition remained the same with prolonged isolated aPTT ranging between 51ã59 seconds. Several years ago, the patient had been tested for anemia and presented a normal coagulation profile at the time. In the absence of extensive hematoma and other bruises, no further investigation was carried out for the abnormally prolonged aPTT.
During the 4th week of the illness, he developed new bruises on both of his hands. There was bleeding from venepuncture site, which did not stop spontaneously. However, there were no changes in the leg hematoma, and the repeated aPTT values still remained high.
On family medicine specialist review, he was referred to hematological team based on clinical suspicion of acquired hemophilia. Upon admission, the patient was actively bleeding from the venepuncture site. Mixing studies with the patientãs plasma were performed but the prolonged aPTT failed to correct completely. Further, the patient had a low plasma factor VIII level of 5%. The plasma VIII inhibitors were also detected, and the titre value was found to be 19.6 BU/mL. Lupus anticoagulant was not detected in patientãs plasma (Tables 1, 2).
Following the diagnosis of acquired hemophilia, the patient was subjected to immunosuppressive therapy in the form of high dose prednisolone 1 mg/kg per day (total 70 mg daily) and cyclophosphamide (100 mg once a day). He was later treated with rituximab (375 mg/m2 on a weekly basis) for a 4-week duration. The patient was also given a short period of inpatient treatment for rehabilitation and rest, allowing the bleeding to stop and hematoma to be resolved. Finally, he was discharged when his aPTT was corrected to normal range.
DISCUSSION
Acquired hemophilia is a rare spontaneous autoimmune disorder caused by the development of antibodies against its own plasma coagulation factors, most commonly factor VIII [1,3]. Acquired hemophilia patients are distributed worldwide, with the incidence of 1.48 per million persons per year [5]. It typically affects the middle-aged group (60ã80 years old) and is reported to occur at equal rates in both sexes [1]. Numerous conditions can give rise to this disorder; however, approximately 50% cases are idiopathic [1]. More severe forms of hemophilia have been observed predominantly in male patients [3].
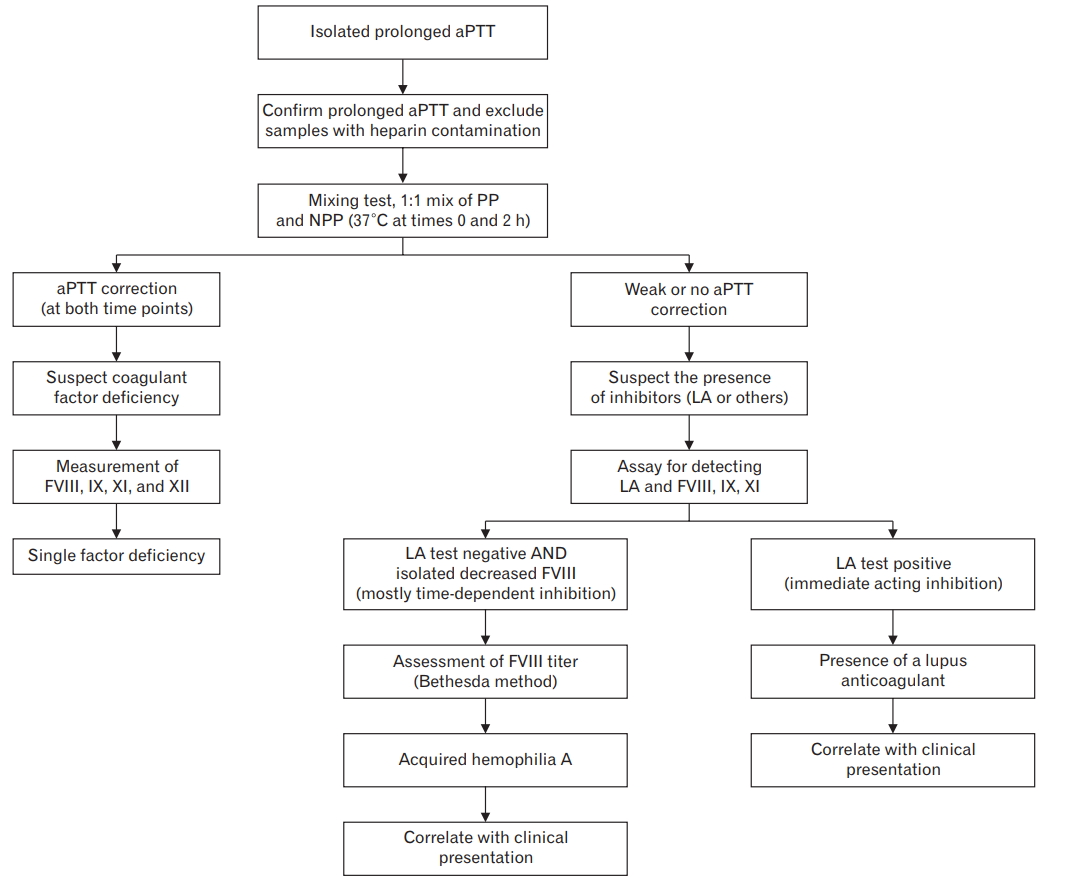
Diagnosis of acquired hemophilia can be challenging. Relative to congenital type, presentation of acquired hemophilia is quite distinct wherein the common manifestation is bleeding into deep soft tissue (hematoma) or purpura, and it rarely involves joints [3]. In our case, the sudden presence of spontaneous spontanous hematoma in an elderly patient without a history of bleeding disorder or trauma should have raised high clinical suspicion for acquired hemophilia. However, it was uncorrected aPTT from plasma mixing test that confirmed the diagnosis of acquired hemophilia. Excluding lupus anticoagulant test results, reduced factor VIII levels and the presence of factor VIII inhibitor were critical for confirming the diagnosis of acquired hemophilia A (Figure 1) [3,6].
The disease is potentially life-threatening with mortality in the range of 8%ã22%, and the patients are at highest risk in the first few weeks following the initial clinical presentation. Mortality remains high among the elderly population, with a rate of 50% at 1 year following diagnosis [2,7].
Treatment of acquired hemophilia includes immediate hemostasis and immunosuppression, and this helps in controlling the autoantibody production. The usual treatment involves administration of a high dose of oral prednisolone at 1 mg/kg combined with oral cyclophosphamide 50ã100 mg per day for up to 2 months with regular aPTT monitoring [3]. Most patients respond well to such combination treatment regime, and monitoring of aPTT is sufficient to study the effectiveness of the treatment in patients. It has recently been shown that treatment of acquired hemophilia with rituximab is more effective and well tolerated. It has also been found to be effective in cases where the conventional treatment is either not responsive or recommended [4]. Treatment with rituximab involves the intravenous infusion of 375 mg/m2 in each administration at weekly interval for a total of 4 weeks [3]. Treating patients with this drug has shown promising results, with an increase in plasma factor VIII levels and a corresponding fall in inhibitor titer within the first week of treatment.
From the current study, it is evident that at initial presentation, diagnosis of acquired hemophilia is not given a priority; further, the isolated prolonged aPTT finding is often ignored. It is critical for the primary health care provider to perform a complete assessment and be aware of rare yet clinically relevant diseases. Considerable attention should be paid while examining the patient, and appropriate investigations should be carried out without any delay. Implications of under-recognized or delayed diagnosis of such diseases can be disastrous to the patient. Early treatment of diseases, such as acquired hemophilia, can significantly reduce the mortality and morbidity resulting from such conditions, particularly for those effective treatments is available.