INTRODUCTION
Primary cutaneous lymphomas (PCL) are a heterogeneous group of skin malignancies of T-cell, B-cell, and, rarely, natural killer-cell origin [1,2]. They present primarily on the skin, without evidence of extracutaneous involvement. PCL with T-cell origin, known as cutaneous T-cell lymphomas (CTCL), constitute a vast majority of PCL cases, with mycosis fungoides (MF) being the most prevalent CTCL [1,3]. MF is a low-grade non-Hodgkin lymphoma wherein the skin is infiltrated with neoplastic T-cells, thereby resulting in the formation of classical skin lesions, ranging from patches to plaques, and later transformation to cutaneous tumors [1-3]. However, patients can present with atypical cutaneous manifestations of MF such as hyperkeratotic, bullous, granulomatous, pustular, and verrucous lesions [3-5]. These diverse manifestations make the diagnosis of MF unquestionably challenging.
Recently, the distinct presentation of MF with pityriasis lichenoides (PL)-like lesions exhibiting recurrent crops of erythematous scaly papules has been reported [4,6-8]. These lesions can lead to widespread hypopigmentation in dark-skinned individuals, thereby masking the characteristic lesions and resulting in misdiagnosis and poor treatment [6]. This new subtype of MF is called pityriasis lichenoides-like MF (PL-like MF). Its clinical findings are similar to those of PL (benign reactive papulosquamous lesions), while its histopathological findings are similar to those of neoplastic MF [7]. PL-like MF is a very rare variant of MF, with only a few cases reported. The first case series was described by Ko et al. [5] in 2000, followed by few authors thereafter [4,5,7,8]. We thereby report a rare case of PL-like MF to raise awareness among primary care physicians regarding the importance of clinical suspicion of MF in children with persistent or worsening papular skin lesions, not responding to standard therapy, to ensure timely referral, diagnosis, and treatment.
CASE REPORT
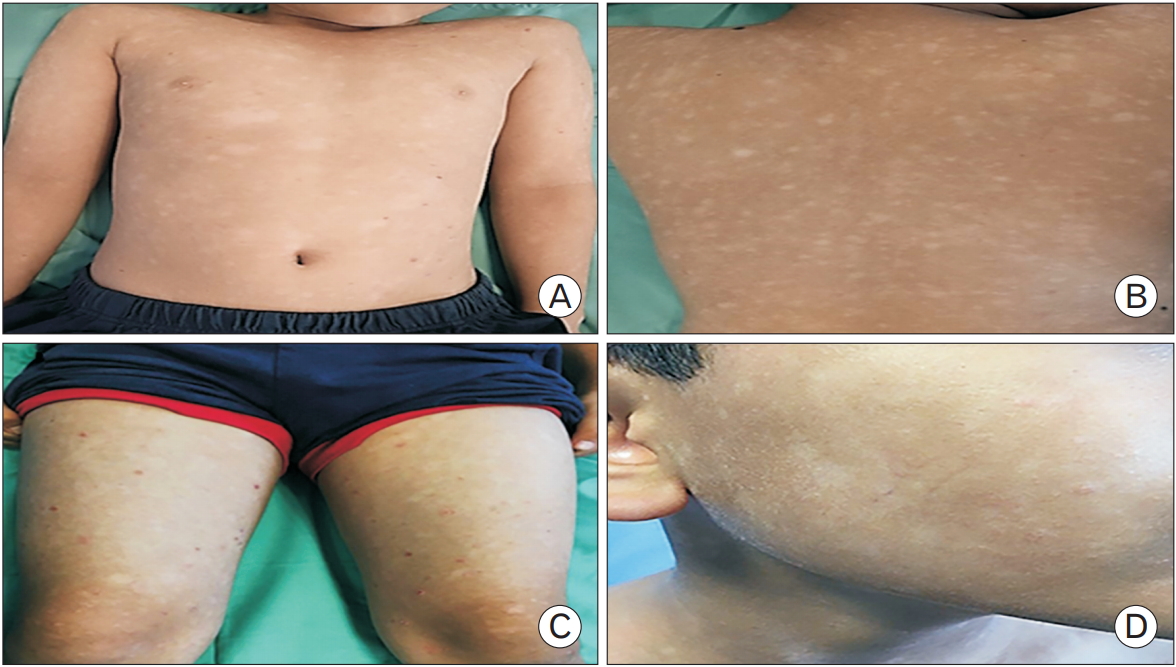
An 11-year-old boy presented with a 2-year history of dermatosis, characterized by multiple scaly erythematous lesions with extensive hypopigmentation. The initial lesions began as erythematous papular eruption on the trunk, and they later spread to the back, limbs, neck, and head. The lesion subsequently flattened and regressed over time, leaving the skin with hypopigmented macules covering almost his entire body (Figure 1). On examination, numerous erythematous discrete papules with fine scales were seen within the area of hypopigmentation (Figure 2). The hypopigmented macules were neither painful nor numb. The patient was otherwise healthy, with no significant past medical history. There was no other history of skin disease or atopy, recent infection, or relevant environmental exposure. Further examination revealed no hepatosplenomegaly and lymphadenopathy. Results of laboratory tests including full blood counts with differential tests as well as renal and hepatic functions were all within the normal range.
Retrospectively, the patient had visited seven clinics for medical treatment, but his skin condition persisted and progressively worsened. The initial stage of pruritic erythematous papules had evoked the diagnosis of early varicella infection. Subsequently, he was treated for atopic dermatitis and pityriasis rosea with various topical emollients and steroids, with no significant improvement. Later, several diagnoses such as tinea corporis, pityriasis alba, pityriasis versicolor, and post inflammatory hypopigmentation were made for the progressive hypopigmented lesions. Antifungals and steroids were also prescribed, but none effectively cured the condition. Then, he was referred to a dermatologist for expert evaluation.
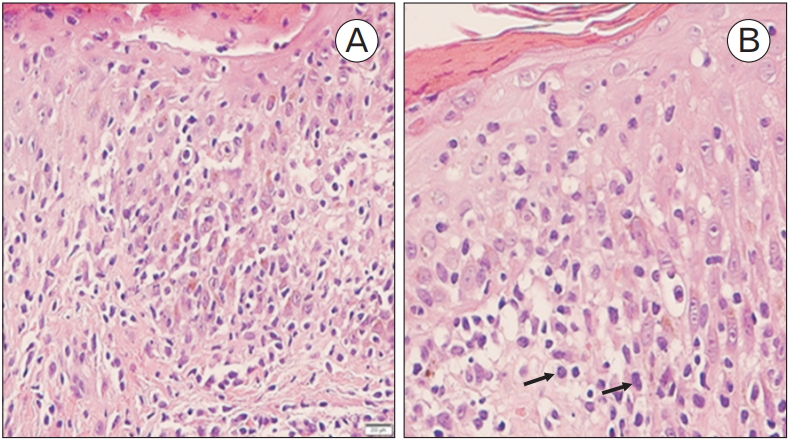
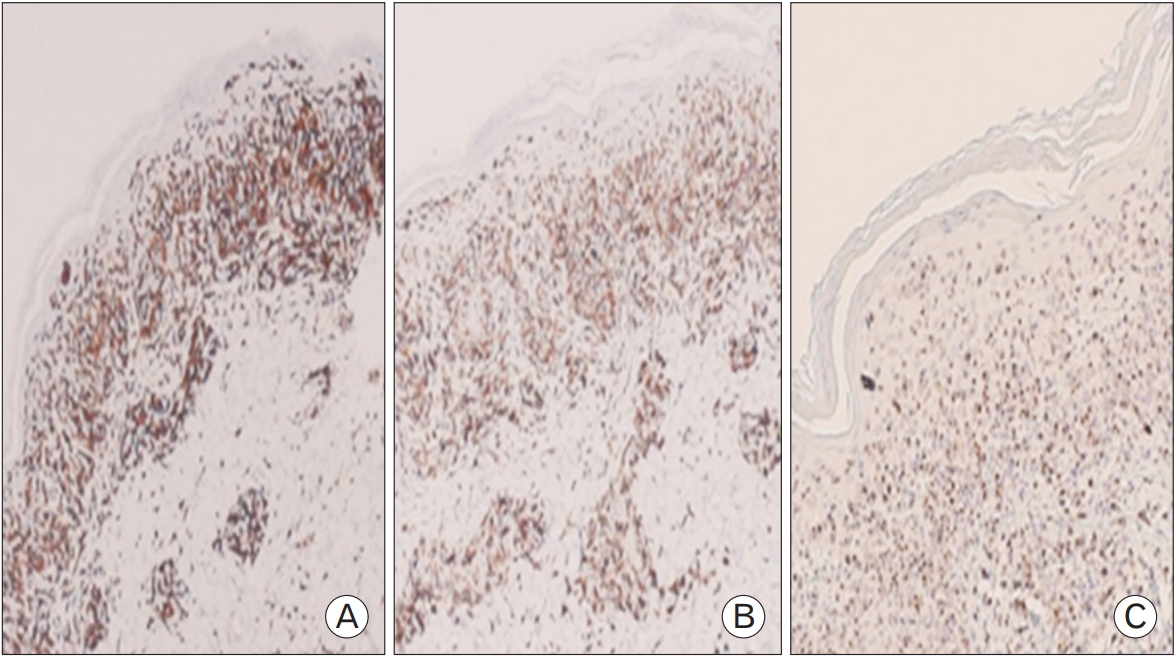
A 4.0-mm punch biopsy was taken from two sites: an erythematous papule and a hypopigmented macule. Histopathological studies on the erythematous papule revealed marked acanthosis and mild parakeratosis with prominent epidermotropism of the dermis. The lymphocytes were atypical in appearance, some of which appeared cerebriform. Pautrier’s microabscess was occasionally seen. The papillary dermis had dense band-like lymphocytic infiltrates, with perivascular lymphocytic and histiocytic cells in the deep dermis (Figure 3). Immunohistochemical staining demonstrated positivity for CD2+, CD3+, and CD5+ and negativity for CD30+ T-cells. CD8+ T-cell expression was more predominant than that of CD4+ T-cells (Figure 4). T-cell receptor gamma gene rearrangement analysis was not performed in this case. Otherwise, biopsy of the hypopigmented macules showed features of post-inflammatory hypopigmentation.
In summary, our patient had clinical presentation of pityriasis lichenoides-like lesions exhibiting recurrent crops of erythematous and scaly papules, which then resulted in prominent post-inflammatory hypopigmentation. Nevertheless, the histopathology combined with immunochemical findings were consistent with those of MF. Therefore, the diagnosis of PL-like MF was established. Treatment thrice weekly with narrowband ultraviolet B (NBUVB) phototherapy, along with topical betamethasone valerate cream 0.05% once daily, was commenced. Significant clinical improvement was observed after completion of 30 phototherapy sessions. A long-term periodic follow-up was planned in order to detect any early symptoms and signs of relapse. The patient provided written consent for the publication of clinical details and images.
DISCUSSION
Pityriasis lichenniodes (PL) and MF are traditionally regarded as two different entities, representing the most common benign and malignant lymphoproliferative disorders in children, respectively [9,10]. PL is characterized by reactive cutaneous eruptions of erythematous scaly papules, while MF has a wide range of clinical presentations depending on its variants. Histopathologic findings of PL include necrotic keratinocytes, spongiosis, erythrocyte extravasation, and perivascular cell infiltration [4,7]. In MF, the remarkable features are Pautrier’s microabscess, epidermotropism, haloed lymphocytes, and coarse collagen bundles in the papillary dermis [4,5]. Recent evidence suggested a relationship between PL and MF, categorized into three groups: PL with T-cell infiltration, PL evolving to MF, and PL-like MF [7-10]. PL-like MF is a rare subtype of MF, described as causing similar benign skin lesions as those of PL but having histopathological findings as those of malignant MF, as illustrated in our case.
The clinical pattern of the PL-like lesion, which is scaly erythematous papules, can be easily mistaken for other more common benign skin conditions. In our case, the patient first arrived at the clinic after having pruritic erythematous papule eruption for 2 days, and the provisional diagnosis of varicella infection was made. However, the absence of vesicles, slow progression, and lack of prodromal symptoms did not further support this diagnosis. Other diagnoses made by different primary care physicians were atopic dermatitis and pityriasis rosea in view of the papulosquamous rashes. Therefore, the patient was treated with various topical emollients and steroids but showed no significant improvement.
Over several weeks, some erythematous lesions slowly faded, and hypopigmented macules and patches started to appear. Primary care physicians face significant dilemma in the diagnosis of hypopigmented skin lesions because the differential blood tests are vast. When the patient presented with fewer hypopigmented macules, the diagnosis was most likely considered as pityriasis alba. Other diagnoses made were superficial fungal infection including tinea corporis, pityriasis versicolor, and post-inflammatory hypopigmentation. Fungal infection can be easily excluded by microscopic examination of skin scrapings in potassium hydroxide solution, but unfortunately, none of the primary care physicians performed the test on this patient. He was treated clinically with topical and oral antifungal medications for a few months, but the hypopigmentation worsened, affecting almost his entire body. Differential diagnoses of vitiligo and leprosy must also be considered in hypopigmentation, but these were very unlikely in this case.
In dark-skinned individuals, the risk for post-inflammatory hypopigmentation after any cutaneous inflammation significantly increases [6]. In this case, widespread hypopigmentation after the resolution of erythematous papules occurred, covering almost his entire body. Extensive hypopigmentation also caused primary care physicians to disregard the residual erythematous lesions, hence only formulating diagnoses for hypopigmentation. A detailed history including the progression of skin lesions, response to previous treatments, and a thorough physical examination must be performed to avoid misdiagnosis and improper treatment. If the skin lesion improves minimally, persists, or worsens, then primary care physicians should refer the case for expert evaluation to avoid undue delay in making a definitive diagnosis. A skin biopsy is often helpful in diagnosing suspicious skin lesions.
In conclusion, PL-like MF is an uncommon and challenging diagnosis not only for primary care physicians but also for dermatologists and pathologists. It is a rare malignant cutaneous disease with overlapping features of benign inflammatory disorders and conflicting clinicopathological findings. This condition should also be taken into consideration during suspected diagnosis in children with persistent or worsening skin lesions, not responding to standard topical treatment. Appropriate referral for expert evaluation and skin biopsy will ultimately lead to timely diagnosis and better outcome. The first-line treatment for children with PL-like MF is phototherapy with NBUVB, and a favorable response can be anticipated [5,8].